Mase-phi HPC
Sep 1, 2024
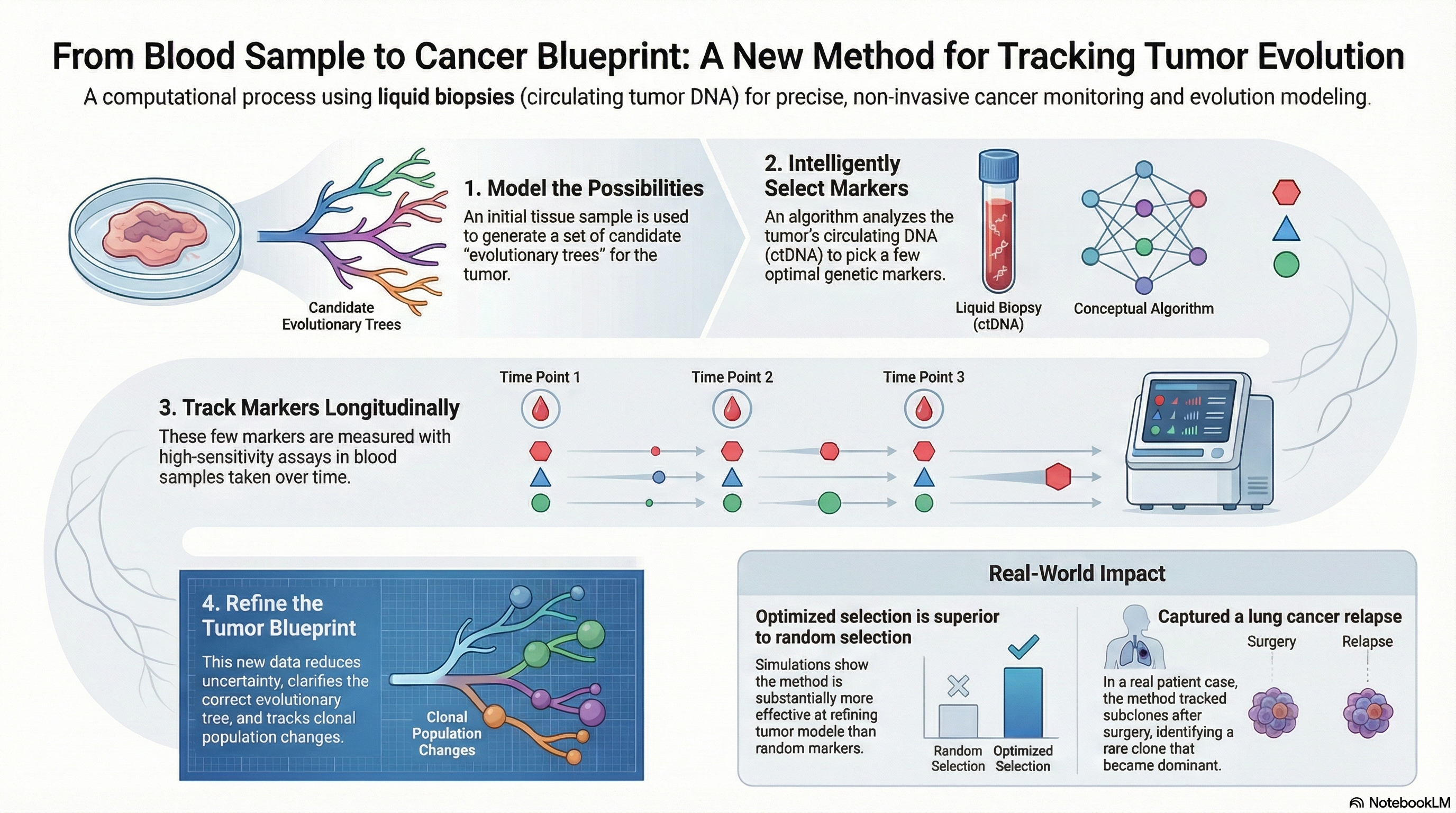
The Mase-phi framework provides a methodology to track a patient's cancer using clonal lineages. This was a successful prototype method, but practical use cases require significantly larger cohorts to be processed. I scaled this method into a high performance computing tool which analyzes a patient in a matter of hours instead of days through joining parallel and concurrent steps. A high performance system for tracking cancer with blood draws.